

Preparing Medical Devices for FDA 510(k) Submission and Manufacturing Transfer
Regulatory compliance is not a final checkpoint but a continuous engineering discipline that begins at the concept phase. According to recent industry data, approximately 80% of medical device companies face delays in their 510(k) submissions due to inadequate design history file (DHF) documentation or misaligned manufacturing processes. This statistic highlights a critical reality: regulatory success is determined by the rigor of your engineering infrastructure, not just the final test results. When teams treat compliance as an afterthought, they risk costly redesigns, extended time-to-market, and potential rejection from the FDA. At A65 Consulting, we have supported more than 8 FDA 510(k) submissions, ensuring that products meet stringent regulatory standards while maintaining engineering integrity. This guide outlines the essential steps to prepare your medical device for a successful submission and seamless manufacturing transfer. (Contact A65 Consulting)
Understanding 510(k) Pathways and Regulatory Strategy
The first step in preparing a medical device for the U.S. market is determining the appropriate regulatory pathway. The 510(k) premarket notification is the most common route for devices that can demonstrate substantial equivalence to a legally marketed predicate device. This process requires a detailed comparison of your device's intended use and technological characteristics against existing products.
Regulatory strategy must be established early in the development lifecycle. According to the FDA, the average time to review a 510(k) submission has increased in recent years, making early engagement with regulatory experts crucial. A65 Consulting helps clients navigate these complexities by identifying the correct predicate and defining the necessary testing protocols from the outset. This proactive approach minimizes the risk of "additional information" requests, which can delay approval by months.
Key considerations include:
- Predicate Selection: Identifying a device with the same intended use and similar technological characteristics.
- Substantial Equivalence: Demonstrating that your device is as safe and effective as the predicate.
- Regulatory Classification: Confirming the device falls under a 510(k)-exempt or 510(k)-required classification.
Implementing Design Controls and the DHF
Design controls are the backbone of any successful 510(k) submission. The FDA requires manufacturers to establish and maintain procedures for design planning, input, output, review, verification, validation, and transfer. These controls ensure that the device meets user needs and regulatory requirements.
The Design History File (DHF) is the primary documentation repository for these activities. It must contain or reference all records necessary to demonstrate that the design was developed in accordance with the approved design plan. A comprehensive DHF includes:
- Design Plan: Outlining the stages of development and responsibilities.
- Design Inputs: Functional, performance, and safety requirements.
- Design Outputs: Drawings, specifications, and manufacturing instructions.
- Design Reviews: Formal evaluations of design progress at critical stages.
At A65 Consulting, we emphasize that the DHF is not just a documentation exercise but a living record of engineering decisions. Our team ensures that every design change is traceable, verified, and validated, providing a clear audit trail for regulatory reviewers. This meticulous attention to detail is what separates compliant devices from those that face regulatory hurdles.
Verification and Validation: Proving Safety and Efficacy
Verification and validation are distinct but complementary processes that prove your device works as intended. Verification confirms that the design outputs meet the design inputs. In simpler terms, it answers the question: "Did we build the device right?" Validation, on the other hand, confirms that the device meets the user needs and intended use. It answers: "Did we build the right device?"
According to industry benchmarks, verification failures are rarely surprises. When a test fails late in development, teams often treat it as an execution issue. However, the underlying problem was likely introduced earlier in the design phase. A65 Consulting advocates for a verification strategy that is integrated throughout the development process, not just at the end. This includes:
- Biocompatibility Testing: Ensuring materials are safe for patient contact.
- Software Validation: Verifying that software functions correctly and securely.
- Usability Testing: Confirming that the device is intuitive and safe for clinical use.
Our engineering team works closely with your quality assurance group to develop robust test protocols that satisfy both regulatory requirements and engineering rigor. This collaborative approach ensures that your device is not only compliant but also reliable in real-world clinical environments.
Manufacturing Transfer and Process Validation
Manufacturing transfer is the process of moving the device design from the development phase to production. This step is critical because it ensures that the device can be manufactured consistently at scale. According to recent regulatory trends, manufacturing issues are a leading cause of 510(k) rejections, highlighting the need for early planning in this area.
Process validation is a key component of manufacturing transfer. It involves documenting clear evidence that a process consistently produces a product meeting its predetermined specifications. This typically includes:
- Installation Qualification (IQ): Verifying that equipment is installed correctly.
- Operational Qualification (OQ): Confirming that equipment operates as intended.
- Performance Qualification (PQ): Demonstrating that the process produces consistent results.
A65 Consulting specializes in bridging the gap between design and manufacturing. We help clients develop manufacturing processes that are scalable, cost-effective, and compliant with FDA standards. Our expertise in design for manufacturing (DfM) ensures that the device is designed with production in mind, reducing the risk of costly redesigns during the transfer phase.
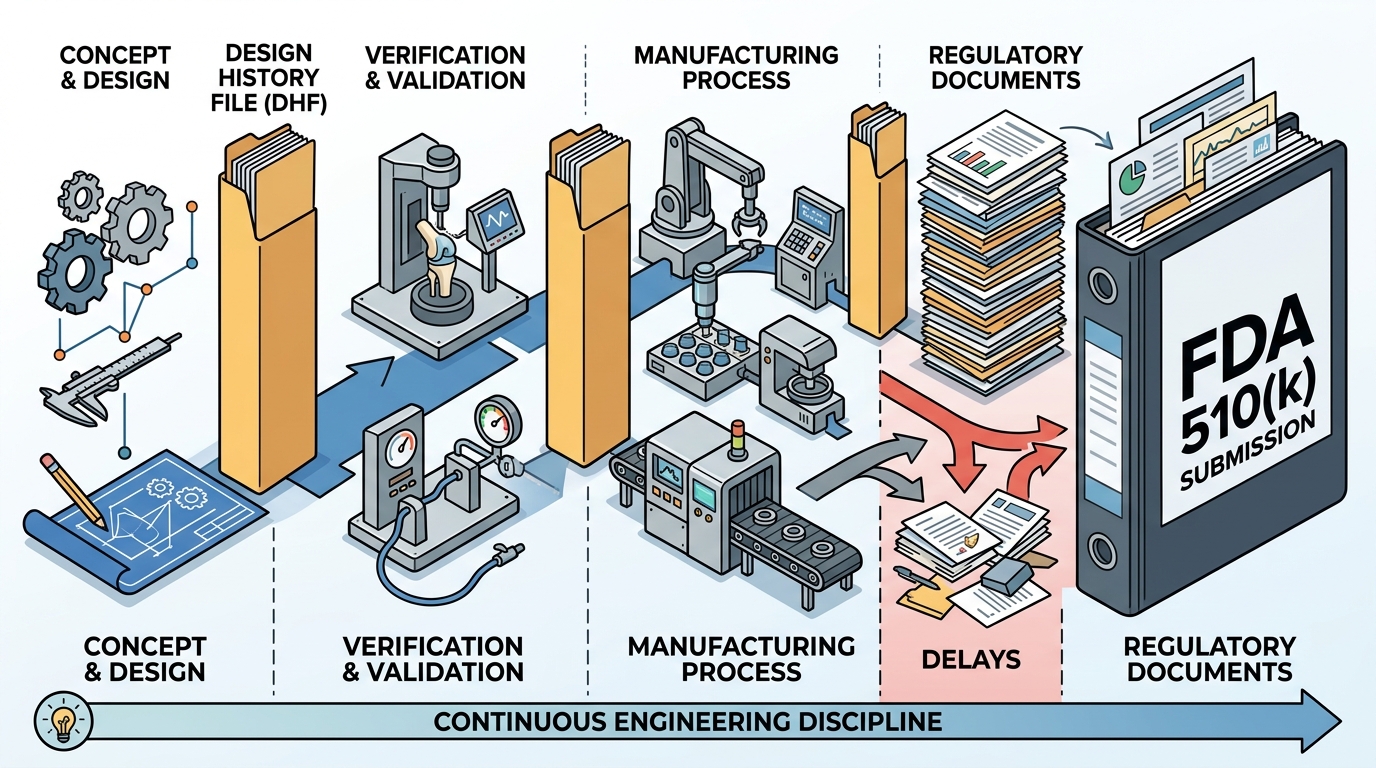
Aligning with Quality System Regulations (QSR)
The FDA's Quality System Regulation (QSR), now transitioning to the Quality Management System Regulation (QMSR), sets the standard for how medical devices are designed, produced, and distributed. Compliance with QSR is mandatory for all 510(k) submissions. This regulation covers all aspects of the quality management system, including management responsibility, design controls, document controls, and corrective and preventive actions (CAPA).
Integrating QSR requirements into your development process from the beginning is essential. This includes:
- Document Control: Ensuring that all documents are approved, reviewed, and updated as necessary.
- Supplier Management: Verifying that suppliers meet quality standards.
- Internal Audits: Conducting regular audits to identify and correct non-conformities.
At A65 Consulting, we help clients build robust quality management systems that not only meet regulatory requirements but also drive operational excellence. Our team provides expert support in developing quality procedures, conducting internal audits, and preparing for FDA inspections. This comprehensive approach ensures that your device is ready for market launch without regulatory surprises.
Key Takeaways
- Early Regulatory Strategy: Identify the correct 510(k) pathway and predicate device early in the development process to avoid delays.
- Comprehensive DHF: Maintain a detailed Design History File that traces every design decision from input to output.
- Integrated Verification: Treat verification as a continuous process, not a final phase, to catch issues early.
- Process Validation: Validate manufacturing processes to ensure consistent, high-quality production at scale.
- QSR Compliance: Align all development activities with FDA Quality System Regulations from day one.
- Expert Partnership: Leverage external engineering expertise to fill skill gaps and accelerate time-to-market.
- Proactive Risk Management: Address potential regulatory and manufacturing risks before they become critical issues.
Frequently Asked Questions
How long does the 510(k) submission process take?
The FDA aims to review 510(k) submissions within 90 days, but the actual timeline can vary based on the complexity of the device and the quality of the submission. Preparing a comprehensive and well-documented submission can significantly reduce review times.
What is the difference between verification and validation?
Verification confirms that the device meets its design specifications ("Did we build it right?"), while validation confirms that it meets user needs and intended use ("Did we build the right device?"). Both are essential for regulatory approval.
Why is manufacturing transfer important for 510(k) approval?
Manufacturing transfer ensures that the device can be produced consistently and safely at scale. The FDA requires evidence that the manufacturing process is validated and controlled to ensure product quality and patient safety.
Can A65 Consulting help with FDA inspections?
Yes, A65 Consulting provides expert support for FDA inspections, including preparation, documentation review, and on-site assistance. Our team has extensive experience navigating regulatory audits and ensuring compliance.
What are the key components of a Design History File (DHF)?
A DHF includes the design plan, design inputs, design outputs, design reviews, verification and validation reports, and design transfer records. It serves as the primary evidence that the device was developed in accordance with regulatory requirements.
How does A65 Consulting charge for its services?
A65 Consulting typically charges on a monthly retainer basis and works toward mutually agreed upon project milestones. This flexible approach allows clients to scale resources as needed throughout the development lifecycle.
What is the role of human factors in 510(k) submissions?
Human factors engineering ensures that the device is safe and effective for its intended users. Usability testing is a critical component of the 510(k) submission, demonstrating that the device can be used correctly in real-world clinical environments.
Ready to Accelerate Your Medical Device Launch?
Navigating the complexities of FDA 510(k) submission and manufacturing transfer requires expertise, precision, and a proactive approach. A65 Consulting is your trusted partner in transforming device concepts into viable, compliant products. With decades of experience and a proven track record of successful submissions, we help you overcome engineering challenges and accelerate time-to-market.
Don't let regulatory hurdles slow down your innovation. Book a discovery call with our team today to discuss your project and explore how we can support your journey from concept to launch.